

Development of novel, alternative, facile, ecofriendly, high yield synthetic process for prazosin
- *Corresponding Author:
- D. A. Patil
Department of Pharmaceutical Chemistry, H. R. Patel Institute of Pharmaceutical Education and Research, Karwand Naka, Shirpur - 425 405, Dhule, Maharashtra, India
E-mail: dilipapatil@gmail.com
Date of Received: 26-03-2010
Date of Modified: 28-09-2010
Date of Accepted: 28-10-2010
Available Online: 15-11-2010
Abstract
Industrial chemistry in the new millennium is widely adopting the con-cept of “Green chemistry” to meet the fundamental scientific challenges. Antihy-pertensive drugs include several of the most widely prescribed drugs like diuretics, beta-blockers, ACE inhibitors, calcium channel blockers, and α-1 adrenorecep-tor blockers. The discovery of prazosin, with very high index of α1/α2 affinity has triggered off a renaissance of interest in αα1-adrenoceptor antagonist drugs for treat-ment of hypertension. The three reported routes for synthesis and manufacture of the α-adrenoceptor antagonist- prazosin had some disadvantages. In present study we had developed new methods for the synthesis of prazosin by using microwave. The most important aspect is the overall yield of this process was ~25 % higher than the other reported methods excluding the use of banned substances.
Keywords
prazosin, green chemistry, process development.
Introduction
Diseases of the arterial tree caused more premature deaths than all other diseases such as cancer and infections combined. Among the major risk factors for arterial diseases, high blood pressure has been identifi ed as the most powerful one [1]. Lowering blood pressure in hypertensive patients requires not only a broad choice of eff ective and well tolerated medications, but also skills to motivate them to comply with the lifelong treatment.
Antihypertensive drugs include several of the most widely prescribed drugs like diuretics, beta-blockers, ACE inhibitors, calcium channel blockers, and α-1 adrenoreceptor blockers. Th ese drugs are also used for other indications including heart failure, ischemic heart disease and renal disease. Th e most widely used antihypertensive drugs fall into six mechanistic “classes”. To treat hypertension more than 100 chemical entities are marketed internationally. Th e number of patients existing for every class of antihypertensive drugs is nearly hundreds and thousands of individual chemical entities are identifi ed to treat hypertension.
Prazosin (a) and its analogs, Fig. (1), or congener drugs namely, Terazosin (b), Doxazosin (c), Alfuzosin (d), Bunazosin (e), and Quinazosin (f) have achieved signifi cant usage as selective α1- adrenoreceptor antagonist drugs useful for the treatment of ‘primary’ or ‘essential hypertension,’ the most common of the cardiovascular disease of modern times and which accounts for nearly 40% of the deaths worldwide [2,3].

Figure 1: Structure of Lead Molecules (Prazosin (a), Terazosin (b), Doxazosin (c), Alfuzosin (d), Bunazosin (e), Quinazosin (f)).
As the logical approach for eff ective management of the arterial hypertension, prazosin and its analogs, with a strong vasodilatory action on the arteriolar vascular bed are used widely. Th ey had many advantages like dictating both resistance and capacitance of blood vessels, favorable haemodynamic eff ects, virtual absence of refl ux tachycardia, maintenance of renal blood fl ow and glomerular fi ltration rate and intact auto regulation of noradrenaline due to non-blockade of presynaptic α2-adrenoceptors. Th ey also had favorable eff ect on lipid profi le, carbohydrate metabolism and insulin resistance. Th ese drugs are also useful in the treatment of congestive heart disease (CHD), variant or prinzmetal’s angina, raynaud’s disease etc. However, there is a need of developing more specifi c analogs with specifi c affi nities to α1-adrenoceptor subtypes to remove their side eff ects [3].
Prazosin has achieved signifi cant clinical usage in the country and has been included in the Indian Pharmacopoeia 1996. However, there are very few Indian manufacturers of prazosin (only one), terazosin (two) and doxazosin (three), which include all Hyderabad based companies namely, Dr. Reddy’s holding (Trident), Aurobindo Pharma, Heterodrugs and SMS Pharmaceuticals. No Indian manufacturer for Bunazosin, Trimazosin and Alfuzosin is listed. A large number of Indian formulators import these drugs. Th e currently used processes[4,5] for prazosin analogs suff ered from many drawbacks like low yields, use of thiophosgene, drastic reaction conditions, high Raw Material Cost (RMC) etc. Th e reported routes for the synthesis and manufacture as depicted below had some disadvantages like Fig. (2);

Figure 2: Reported routes for synthesis of Prazosin
Route I: Overall yields 8-10%, use of Th iophosgene (toxic and banned substance which had carcinogenic property).
Route II: Very low overall yields, drastic reaction conditions and prolonged reaction time required to complete the reactions.
Route III: Longer synthesis of multiple steps, which increased the overall cost of the product.
Th ere was a need and opportunity to develop an alternative, cost eff ective, facile and eco-friendly route for the synthesis of prazosin and its congeners.
Materials and Methods
Source of chemicals and apparatus
Reagents used in the synthesis were of analytical or laboratory grade and were used as supplied or were prepared according to procedures described in literature. Solvents except analytical reagent grade were dried and purifi ed according to literature when necessary. Reactions were monitored by thinlayer chromatography (TLC) on pre-coated silica gel F254 plates from Merck and compounds visualized by exposure to UV light. Melting points were determined in open capillary with Veego electronic apparatus and are uncorrected.
Th e IR spectra were recorded using potassium bromide on JASCO V-5300 FT-IR. Th e 1H NMR spectra were recorded in CDCl3 and DMSO using NMR Varian-Mercury 300 MHz spectrometer. Chemical shifts are reported in δ ppm units with respect to Tetramethyl Silane as internal standard and coupling constants (J) were reported in Hz units. Mass spectra were obtained on an Electron Impact mass spectrometer at 70 eV ionizing beam using direct insertion probe.
Synthesis
Th e starting material 3,4-dimethoxybenzaldehyde[6] and Methyl-2-amino-4,5-dimethoxybenzoate (a) required for the synthesis of target compound was prepared by the reported method [4].
Synthesis of Bioisoster of Prazosin:
All prime starting materials required in the scheme II had been synthesized, and at this moment it was decided to setup reaction conditions of various steps involved in scheme II. Compared to directly trying the reactions with 4,5-dimethoxyanthranilonitrile or methyl-2-amino-4,5-dimethoxybenzoate which were rather not easily accessible because it can be only prepared by multistep syntheses, it was thought most of process development work could be done with more simple, inexpensive and easily accessible starting material. One such starting material, bioisoster of dimethoxy-methylanthranilate, was 2-amino-3- carbethoxy-4,5,6,7-tetrahydrobenzo-(b)-thiophene (4). It was prepared in a one-pot, single step process in excellent yield by utilizing Gewald reaction [7]. Th us, it was decided to adopt scheme III, involving use of thiophene-o-amino-ester as starting material, which bestow the thiophene analog of prazosin.

Figure 3: (I) NH¬¬¬¬2CSNH2, HCL(50%), MW; (II) dimethyl sulphate, NaOH (20%), 0-5°C, 3-4h; (III) unhydrous piperazine, isopropyl alcohol, MW; (IV) 2-furoyl chloride, isopropyl alcohol, potassium carbonate, TEBA chloride, MW; (V) POCl3, isopropyl alcohol, MW; (VI) NH3, THF, 0-5°C, 2h; (VII) NaN3, THF, 145-150°C; (VIII) Zn, AcOH, 85-90°C.

Scheme III: Bioisosteric analog of Prazosin (I) NH¬¬¬¬2CSNH2, HCL(50%), MW; (II) dimethyl sulphate, NaOH (20%), 0-5°C, 3-4h; (III) unhydrous piperazine, isopropyl alcohol, MW; (IV) 2-furoyl chloride, isopropyl alcohol, potassium carbonate, TEBA chloride, MW; (V) POCl3, isopropyl alcohol, MW; (VI) NH3, THF, 0-5°C, 2h; (VII) NaN3, THF, 135-140°C; (VIII) Zn, AcOH, 95-105°C.
Synthesis of Prazosin bioisoster, 4-amino-2- [(4-(2-furoyl)-1-piperazinyl]-5,6,7,8-tetrahydrobenzo-( b)-thieno-(2-3d)-pyrimidine-4-(3H) (4f); Synthesis of 2-amino-3-carbethoxy- 4,5,6,7-tetrahydrobenzo-(b)-thiophene[7,8] (4):
A mixture of cyclohexanone (1 mol), sulfur (0.1 mol), ethylcyanoacetate (0.1 mol) and ethanol (20 ml) was stirred. To well stirred mixture diethylamine (0.125 mol) was added dropwise for half hour and stirring continued for another 3 hours at ambient temperature, once all sulfur got dissolved, reaction mixture was kept at 0-5°C for overnight. Th e solid separated was fi ltered next day, washed with 20 ml chilled 50% aqueous methanol. Th e product (75% yield) having mp 111-112°C (115°C) was characterized as 2-amino-3-carbethoxy-4,5,6,7-tetrahydrobenzo- (b)-thiophene.
Yield 75%, mp 111-112°C, IR (KBr) cm-1: 3414, 3306, 3165, 3074, 2988, 1649. Anal. Calcd for C11H15NO2S.
Synthesis of 2-Mercapto-5,6,7,8-tetrahydrobenzo-( b)-thieno[2,3-d]pyrimidine-4-one (4a):
A mixture of 2-amino-3-carbethoxy-4,5,6,7-tetrahydrobenzo-( b)-thiophene (0.004mole) and thiourea 0.008mole) were added to a round bottom fl ask and mixed well, to this mixture HCl (50%) was added till moist mass obtained. Th e mixture was irradiated with microwave (90 W) for 5 min, with cold shocks of half min. Th e reaction mixture was dissolved in 30ml sodium hydroxide solution (20% w/v) cooled to 0-5°C. Th e solution was acidifi ed with conc. HCl producing white colored solid, washed with cold water, fi ltered out, and dried. Th e product (87% yield) having mp 212-214°C (213-215°C) was characterized as 2-mercapto-5,6,7,8-tetrahydrobenzo(b) thieno[2,3-d]pyrimidine-one(4a).
Yield 87%, mp 212-214°C, IR (KBr) cm-1: 1802, 1678, 1598, 669. Anal. Calcd for C10H10 N2OS2
Synthesis of 2-thiomethyl-4-oxo-5,6,7,8-tetrahydrobenzo-( b)-thieno[2,3-d]pyrimidine (4b):
2-mercapto-5,6,7,8-tetrahydrobenzo-(b)- thieno[2,3-d]pyrimidin-4one (3a) (0.02mole) was dissolved in 20 ml sodium hydroxide solution (20% w/v), cooled to 0-5°C. In this solution dimethyl sulphate (0.04mole) was added dropwise for half hour with stirring. Th e reaction mixture was further stirred for another 2 h and kept at 0-5°C for overnight. Th e solid separated out was fi ltered, washed and dried. Th e product (67% yield) having m.p.235- 237°C (310-311°C) was characterized as 2-thiomethyl- 5,6,7,8-tetrahydrobenzo(b)thieno[2,3-d] pyrimidin-4-one(4b).
Yield 67%, mp 310-311°C, IR (KBr) cm-1: 2985, 1722, 1657, 1597, 656. Anal. Calcd for C11H12 N2OS2
Synthesis of 2-(N-piperazinyl)-5,6,7,8-tetrahydrobenzo( b)thieno[2,3-d]pyrimidin-4-one (4c):
A solution of 2-thiomethyl-4-oxo-5,6,7,8-tetrahydrobenzo( b)thieno[2,3-d]pyrimidine (0.012mole) and anhydrous piperazine (0.036mole) were mixed in a round bottom fl ask, fi tted with a refl ux condenser. Th is mixture was refl uxed for 5-6 h. Th e reaction mixture was poured in ice-water (30ml) solid separated out was fi ltered washed and dried. Th e product (86% yield) having m.p.214-216°C was characterized as 2-(N-piperazinyl)-4-oxo-5,6,7,8- tetrahydrobenzo(b)thieno[2,3-d]pyrimidine (4c).
Yield 86%, mp 214-216°C, IR (KBr) cm-1: 2958, 1705, 1679, 1685, 1556, 675. Anal. Calcd for C14H18N4OS.
Synthesis of 2-[4-furoyl-(N-piperazinyl)]- 4-oxo-5,6,7,8-tetrahydrobenzo(b)thieno- [2,3-d]-pyrimidine(4d):
A mixture of 2-(N-piperazinyl)-4-oxo-5,6,7,8- tetrahydrobenzo(b)thieno[2,3-d] pyrimidine (4c) (0.017 mol) and 2-furoyl chloride (0.034 mol) was prepared in N,N’-dimethylformamide (50ml) in a round bottom fl ask, then potassium carbonate (0.034mole) and TEBA-chloride (0.001mole) were added. Th e resulting mixture was irradiated with microwave for 2-2.5 h. (90 W), then the reaction mixture was poured in ice cold water and the solid separated out was fi ltered and dried. Th e product (85% yield) having m.p.285-287°C was charaO2cterized as 2-[4-furoyl-(N-piperazinyl)]-4-oxo-5,6,7,8- tetrahydrobenzo(b) thieno[2,3-d]pyrimidine (4d).
Yield 85%, mp 285-287°C, IR (KBr) cm-1: 2959, 1711, 1597, 1597, 1189, 1145, 679. Anal. Calcd for C19H20N4O3S.
Synthesis of 2-[4-furoyl-(N-piperazinyl)]-4- chloro-5,6,7,8-tetrahydrobenzo(b)thieno[2,3-d] pyrimidine(4e):
Th e 2-[4-furoyl-(N-piperazinyl)]-4-oxo-5,6,7,8-tetrahydrobenzo( b)thieno[2,3-d] pyrimidine (4d), (0.01mole) was dissolved in phosporous oxychloride (1.5 mol), in an iodine fl ask fi tted with a refl ux condenser and a guard tube. Th e reaction mixture was refl uxed for 3-4 h. From the reaction mixture phosphorous oxychloride was distilled out at reduced pressure. Th ereafter reaction mass was quenched in ice cold water, and upon neutralization with sodium bicarbonate, precipitate obtained. Th e product (76% yield) having m.p.178-181°C was characterized as 2-[4-furoyl-(N-piperazinyl)]-4-chloro-5,6,7,8-tetrahydrobenzo (b)thieno[2,3-d]pyrimidine (4e).
Yield 76%, mp 178-181°C, IR (KBr) cm-1: 2898, 1781, 1581, 1601, 1139, 1074, 789. Anal. Calcd for C19H19ClN4O2S.
Synthesis of 2-[4-furoyl-(N-piperazinyl)]-4- amino-5,6,7,8-tetrahydrobenzo(b)thieno[2,3-d] pyrimidine (4g):
A solution of 2-[4-furoyl-(N-piperazinyl)]-4-chloro- 5,6,7,8-tetrahydrobenzo(b)thieno[2,3-d] pyrimidine (0.001mole) was prepared in tetrahydrofuran (25 ml) in a round bottom fl ask, and cooled to 0-5°C. Dry ammonia gas was generated and passed through the solution of compound. A white colored precipitate was separated out, which was fi ltered out and dried. Th e product (68% yield) having mp 210-212°C was characterized as 2-[4-furoyl-(Npiperazinyl)] 4-amino-5,6,7,8-tetrahydrobenzo (b) thieno[2,3-d]pyrimidine(4g).
Yield 68%, mp 210-212°C, IR (KBr) cm-1: 3504, 2898, 1781, 1581, 1601, 1139, 1074, 679.
1H NMR (DMSO-d¬¬6) δ: 2.6-2.7 (m, 2H, CH2), 2.85 (m, 2H, CH2), 3.1-3.3(m, 4H, CH2), 3.7-4.1 (m, 8H, CH2), 6.5 (broad s, 1H, CH2), 7.0 (broad s 1H, CH), 7.3 (broad s, 1H, CH), 7.5 (s, 2H, NH2). Anal. Calcd for C19H21N5O2S.
Th e fi nal compound 2-[4-furoyl-(N-piperazinyl)] -4-amino-5,6,7,8-tetrahydrobenzo (b)thieno[2,3-d] pyrimidine can also be obtianed by the reaction of cholro compound (4e) with sodium azide (0.001 mol) in tetrahydrofuran (15 ml) at 135-140°C to produce azido intermediate (4f) with yield of 56%, and this can be further reduced with zinc (0.09 mol) in presence of acetic acid (10 ml) at 95-105°C to aff ord the fi nal compound 2-[4-furoyl-(N-piperazinyl)]4- amino-5,6,7,8-tetrahydrobenzo(b)thieno[2,3-d]pyrimidine (4g) with 45% yield.
Synthesis of Prazosin:
Having synthesized principal starting material required for synthesis of prazosin and also successfully undertaken synthesis of its thieno-(2,3-d)-pyrimidine bioisosteric analog, the actual synthesis of prazosin was now taken up. Synthesis
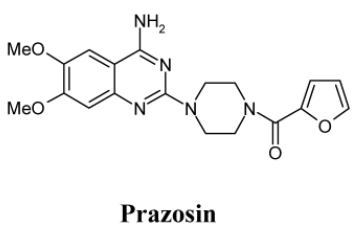
Synthesis of 2-mercapto-6,7-dimethoxyquinazolin- 4-(3H)-one (b):
Th e methyl-2-amino-4,5-dimethoxybenzoate- (0.02mole) (2e), and thiourea (0.04mole), were mixed well and added HCL (50%) till wet mass obtained, in a round bottom fl ask. Th is mixture was irradiated with microwave for 5 min with shocks of 30 sec. Th e reaction mixture was dissolved in 30ml sodium hydroxide solution (20%), and fi ltered to remove any un-dissolved impurities, then cooled to 0-5°C, Th e solution was acidifi ed with conc. hydrochloric acid; the greenish colored solid separated was fi ltered out, washed with ice-cold water, and dried. Th e product (77% yield) having m.p. 245-247°C was characterized as 2-mercapto-6,7-dimethoxyquinazolin- 4-(3H)-one (b).
Yield 77%, mp 245-247°C, IR (KBr) cm-1: 1756, 1702, 1678, 1596, 688. Anal. Calcd for C10H10N2O3S
Synthesis of 6,7-dimethoxy-2-(methylthio) quinazolin-4(3H)-one (c):
Th e 2-mercapto-6,7-dimethoxyquinazolin-4(3H) one (0.006mole) was dissolved in 20ml sodium hydroxide solution (20%), cooled to 0-5°C. To the above solution, dimethyl sulphate (0.018mol) was added drop wise for half an hour with stirring. Again it was stirred for another 2 h and placed in freezer for overnight. Th e solid separated out was fi ltered and dried. Th e product (84% yield) having m.p. 234-236°C was characterized as 2-thiomethyl- 6,7-dimethoxyquinazolin-4(3H)one (c).
Yield 84%, mp 234-236°C, IR (KBr) cm-1: 1747, 1713, 1667, 1597, 658. Anal. Calcd for C10H12N2O3S
Synthesis of 6,7-dimethoxy-2-piperazin-1-ylquinazolin- 4(3H)-one (d):
Solution of 2-thiomethyl-6,7-dimethoxyquinazolin- 4(3H)one (0.003mole) and anhydrous piperazine (0.009mole) was dissolved in isopropyl alcohol in a round bottom fl ask fi tted with a refl ux condenser. Th is mixture was irradiated with microwave for 30- 40min. Th e reaction mixture was quenched in icewater (30ml); solid separated was fi ltered out, and dried. Th e product (82 % yield) having m.p. 325- 327°C was characterized as 6,7-dimethoxy-2-piperazin- 1-yl-quinazolin-4(3H)-one (d).
Yield 82%, mp 325-326°C, IR (KBr) cm-1: 3514, 1757, 1711, 1597, 1597, 1189. Anal. Calcd for C¬14H18N4O3.
Synthesis of 4-furoyl-2-(piperazin-1-yl)-6,7- dimethoxyquinazolin-4(3H)one (e):
A mixture of 2-(piperazin-1-yl)-6,7-dimethoxyquinazolin- 4(3H)one (0.017 mol), and 2-furoyl chloride (0.04mol) was dissolved in isopropyl alcohol (30ml) in a round bottom fl ask, fi tted with a refl ux condenser. To this mixture potassium carbonate (0.03mol) and the PTC namely TEBA chloride in catalytic amount (0.001mole) was added. Th is mixture was irradiated with microwave (90W) (Intermittent cold cycles) for 30min. Th e reaction mixture was quenched in ice-water; solid separated was fi ltered and dried. Th e product obtained in 85% yield was characterized as 4-furoyl-2-(piperazin-1-yl)-6,7- dimethoxyquinazolin-4(3H)one(e). Th us, the crude product was recrystalised from methanol: chloroform (4:1) to aff ord the crystalline product having mp 278-280°C which was characterized by IR.
Yield 85%, mp 278-280°C, IR (KBr) cm-1: 1757, 1711, 1597, 1597, 1189, 1145. Anal. Calcd for C¬19H20N4O5
Synthesis of 2-[4-furoyl-(piperazin-1-yl)]-4- chloro-6,7-dimethoxyquinazoline (f):
Th e 4-furoyl-2-(1-piperazinyl)-6,7-dimethoxyquinazolin- 4-one (0.01mole) was dissolved in isopropyl alcohol (40ml), in an iodine fl ask fi tted with a refl ux condenser and guard tube. Th e reaction mixture was mixed with phosphorus-oxychloride (0.01mol) and refl uxed for 8-9 hrs. Th e reaction mixture was quenched in ice-water and neutralized with sodium bicarbonate; the solid was fi ltered and dried. Th e product (78% yield) having m.p.175-178°C was characterized as 2-[4-furoyl-(1-piperazinyl)]-4- chloro-6,7-dimethoxyquinazoline (f).
Yield 78%, mp 175-176°C, IR (KBr) cm-1: 1787, 1781, 1581, 1601, 1139, 1074, Anal. Calcd for C¬19H19ClN4O4
Synthesis of 2-[4-(2-furoyl)-piperazin-1-yl]-4- amino-6,7-dimethoxyquinazoline (h):
2-[4-furoyl-(piperazin-1-yl)]-4-chloro-6,7-dimethoxyquinazoline (0.009 mol) was dissolved in tetrahydrofuran in a round bottom fl ask, which was cooled at 0-5°C. Th e dry ammonia gas was generated from the gas generation assembly which was passed through the solution of 2-[4-furoyl-(piperazin-1- yl)]-4-chloro-6,7-dimethoxyquinazoline for about 2-3 h. Th e white colored product was separated out, which was fi ltered out, and dried. Th e product (70% yield) was characterized as 2-[4-(2-furoyl)-piperazin- 1-yl]-4-amino-6,7-dimethoxy-quinazoline (g).
Yield 70%, mp 278-280°C, IR (KBr) cm-1: 3504, 1650, 1580, 1528, 1465, 1287, 1000. 1H NMR (DMSO-d¬¬6) δ: 3.2-3.4 (m, 6H, CH2), 3.6-4.1 (m, 8H, Piperazinyl), 6.4-6.45 (br. s, 1H, CH), 6.8- 7.0 (br. s, 1H, CH), 7.8 (s, 1H, CH), 7.5-7.7 (m, 2H, Ar-H), 8.8 (s, 2H, NH2). MS m/z: 383 (M+), 288, 245, 233, 204. Anal. Calcd for C¬19H21N5O4.
Th e molecule 2-[4-(2-furoyl)-piperazin-1-yl]-4-amino- 6,7-dimethoxy-quinazoline can also be obtianed by the reaction of cholro compound (f) with sodium azide (0.001 mol) in tetrahydrofuran (15 ml) at 145- 150°C to produce azido intermediate (g) with yield of 46%, this can be further reduced with zinc (0.09 mol) in presence of acetic acid (10 ml) at 85-90°C to aff ord the fi nal compound 2-[4-(2-furoyl)-piperazin-1-yl]-4- amino-6,7-dimethoxy-quinazoline (h) with 34% yield.
References
- De Smet PA. The role of plant-derived drugs and herbal medicines in healthcare. Drugs. 1997; 54(6): 801-40.
- Brunton LL, Lazo JS, Parker KL. Eds. Goodman and Gilman’s The Pharmacological basis of therapeutics. IX ed. USA. Mc Graw Hill medical publishing division; 1996: 780.
- Manfred EW. Ed. Antihypertensive Agent, Burger’s Medicinal Chemistry and Drug Discovery. Vol.2, Xth ed. Wiley-Interscience publication; 1996: 296-297.
- Honkanen E, Pippuri A, Kairisalo P, et al. New practical synthesis of prazosin. J. Heterocycl. Chem. 1980; 17: 797-798.
- Althius TS, Hess HJ. Synthesis and identifi cation of the major metabolites of prazosin formed in dog and rat, J. Med. Chem. 1977; 20: 146-149.
- Furniss BS, Hannaford AJ, Smith PWG, Et al. Vogel’s textbook of practical organic chemistry, Vth ed. London. Pearson Prentice Hall Low price edition; 2004: 987.
- Gewald K. Angew. Chem. 1961; 73: 114-118.
- Gewald K. Chem. Ber. 1965; 98: 3571-3578.